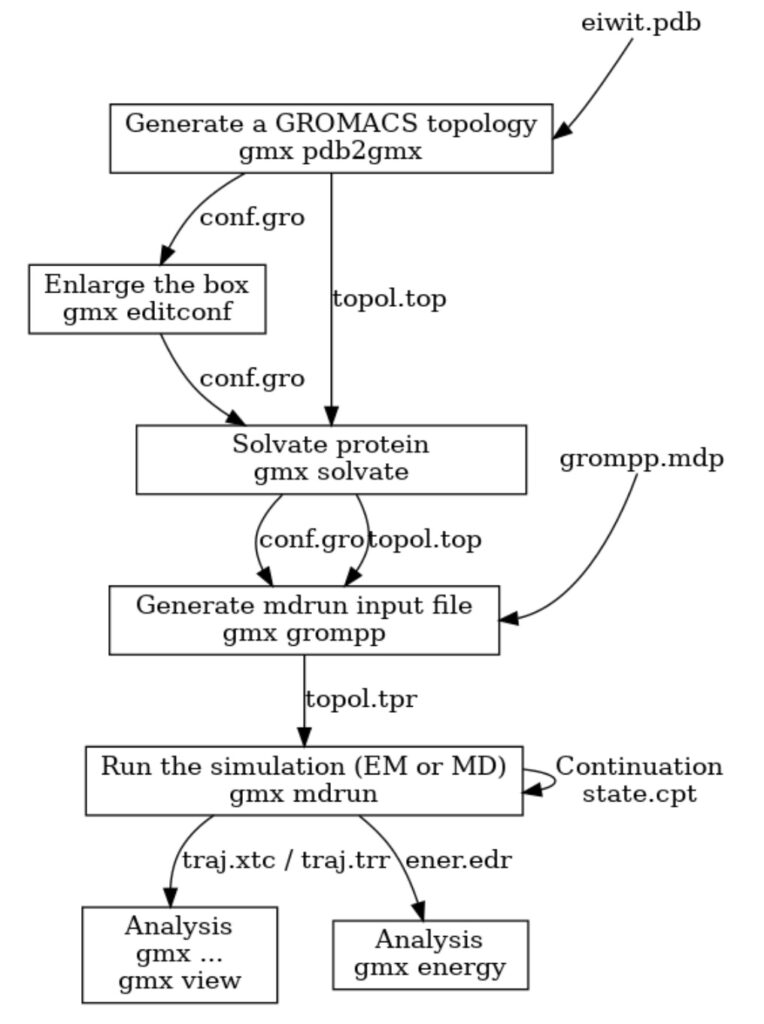
GROMACS has long had an extremely useful preprocessor tool that prepares molecular dynamics simulations for execution. The preprocessor is called with the command gmx grompp (ie. the GROMacs PreProcessor) and it has online documentation. It is a key part of the workflow used workflow used to run a molecular simulation (see Figure 1). Users describe their simulation in text files, separating the description into several parts to promote re-use. Often the same molecular description will be re-run with different algorithms, or the same set of algorithms used for variations of the same set of molecules/force fields, or simply different starting atomic coordinates. Users often do this work on their laptops, and it is convenient to provide warnings to users before they go to the trouble of trying to run a simulation via a batch system. The main output of gmx grompp is a portable binary version of the processed input (called a “.tpr file”). This can be transferred to any computer cluster (regardless of hardware) and the user knows that the simulation will work the same way everywhere.
Much of this code was written over several decades by different people handling different use cases. For example, different force fields describe atomic interactions differently, and require different molecular descriptions to be written and parsed. Almost all of it pre-dates the transition to using C++ and is written in styles that mostly work but are no longer idiomatic. Improving the expression of the code slowly over time is likely to help developers eliminate bugs and provide capability improvements in future. ENCCS has been contributing to this work. There are many useful vocabulary types and algorithms in the C++ standard library that have been found useful in improving the implementation of gmx grompp, including std::vector, std::optional, std::equal, and std::unordered_map.
GROMACS optimizes the code that runs the molecular simulation trajectory very aggressively, because often upwards of trillions of iterations are needed to perform useful science. However, the preprocessing stage happens only once per trajectory, so gets much less love from the developers. However, as we transition to the exascale, complex simulation workflows that run many different simulation trajectories are becoming more common. Any extra time taken to set up simulations from new starting points might lead to extra opportunity cost – wasting time of expensive GPUs lying idle – so it is worth some effort to improve things.
Over 2021, several improvements were made, mentioned in chronological order:
- Paul Bauer of KTH and BioExcel used std::optional to improve how the lookup of parameters for interactions from force field databases was expressed (commit)
- This made some VDW parameter lookups in a loop more expensive, so Berk Hess of KTH and BioExcel lifted the lookups out of the loop over interactions (commit)
- Mark Abraham of UU and ENCCS improved the memory allocation pattern of VDW parameter preparation (commit)
- Mark also simplified some logic to prepare for the changes below (commit)
- Mark also lifted a complex lookup of the atom type of an atom out of a loop that assigned parameters from the force field database (commit)
- Mark avoided some expensive lookup functions that were called for every dihedral interaction when assignment parameters (commit)
- Long ago, Paul replaced an ancient C data structure with a form of map. Mark refactored some other tools that used that map in unnecessarily complex ways, so that the implementation of that map could be replaced by std::unordered_map (commit)
- Finally, Mark eliminated the custom map by using the standard one (commit)
Overall, we expected the code to get faster, as well as simpler and expressed using more standard idioms. How did we do?
I took the six systems used in benchmarking for the recent GROMACS paper (https://doi.org/10.1063/5.0018516, systems available in the supplementary information at https://doi.org/10.5281/zenodo.3893788), as they are indicative of a range of sizes and kinds of simulations commonly run in GROMACS. Each comes with the full inputs needed to run gmx grompp to prepare the .tpr file. Ten versions of GROMACS were compiled with gcc 9 on a 2016-era Kaby Lake Dell laptop running Ubuntu 20.04 and the performance recorded with the time utility. Ten runs were performed, with the arithmetic mean reported. As gmx grompp is not multi-threaded or MPI-aware, no such parallelism affects these comparisons. The code versions were those from the 2021 master branch described above, plus for baseline comparisons the 2021.4 release and the commit from the 2021 master branch that preceded the introduction of std::optional. The comparisons can be seen in Table 1.
| 2021.4 | Baseline | Use optional | Lift lookup out of loop 1 | Optimize VDW allocations | Simplify logic | Lift lookup out of loop 2 | Avoid lookups | Better data structure | Use unordered_map | |
| adh_dodec | 1.92 | 1.90 | 2.05 | 2.02 | 2.03 | 2.08 | 1.18 | 1.01 | 0.99 | 0.99 |
| aqp_ensemble | 2.22 | 2.23 | 2.38 | 2.23 | 2.25 | 2.30 | 1.28 | 1.06 | 1.07 | 1.06 |
| ion_channel | 0.96 | 0.96 | 1.00 | 1.00 | 0.99 | 0.99 | 0.74 | 0.69 | 0.66 | 0.66 |
| rnase_cubic | 0.28 | 0.28 | 0.30 | 0.29 | 0.29 | 0.29 | 0.21 | 0.20 | 0.19 | 0.19 |
| stmv | 37.56 | 37.62 | 40.38 | 39.70 | 39.84 | 40.04 | 31.27 | 30.41 | 30.06 | 30.19 |
| villin | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 | 0.11 | 0.09 | 0.08 | 0.08 | 0.08 |
Clearly the stmv benchmark takes a lot longer than the others. Not only does it have the largest number of particles (around 1 million) but it has the most diversity of different kinds of molecules, each of which is separate work for grompp. Execution time is approximately proportional to atom count, as expected. The changes introduced in each code change (ie table column) are now discussed, in chronological order.
- From the first two columns we can see that little changed in the master branch that affected the performance of gmx grompp between when the 2021 release branch was forked off and when the baseline was measured. This suggests that we didn’t omit to analyze any significant changes that happened during 2021 in the master branch.
- Using std::optional caused a minor increase in run time in most benchmarks. Profiling suggested that the extra work came from the larger return type from the custom map used for PreprocessingAtomTypes, and more work to compare whether that optional value matched other values.
- Berk’s change to lift out the lookup of atom types from the LJ parameter-generation loop had the most impact on the CHARMM36 benchmark (aqp_ensemble)
- Pre-allocating VDW parameter buffers, rather than re-sizing them in the loop, had little effect
- Mark’s simplifications to the logic had little effect but perhaps slowed some of the cases, while preparing for the future improvements
- Mark’s change to lift atom-type lookups out of the loop over bond types had a clear performance improvement for all benchmark cases.
- Mark’s further change to avoid atom-type lookups in some other cases continued to show performance improvements
- Improving the implementation of the map data structure made only minor improvements to performance
- Using std::unordered_map instead of the custom map had no effect.
The noteworthy relative performance changes are shown in Figure 2 below.
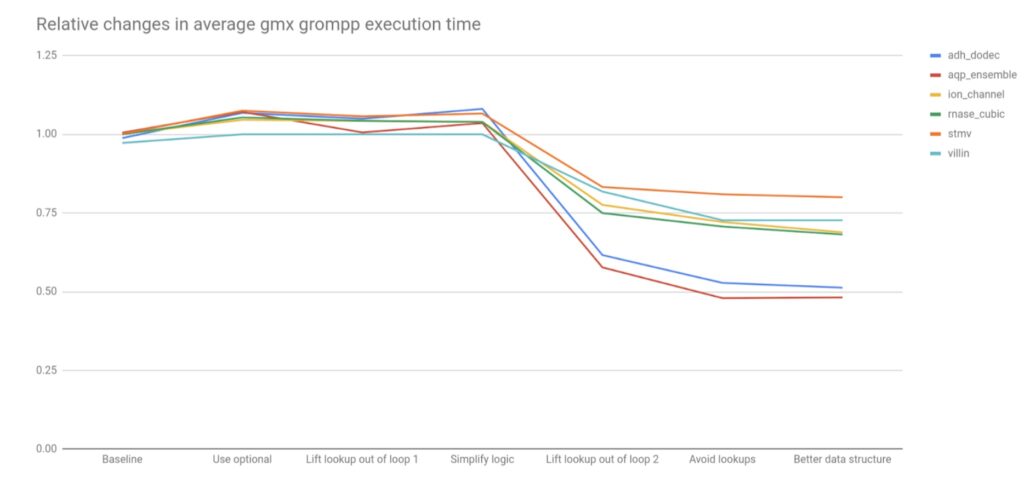
20-50% overall performance improvements on these benchmarks were observed, so it is likely that all GROMACS users will see benefits, starting from version 2022. It is worth noting that several of the refactorings that simplified the code had no significant effect on performance. However these improved non-functional aspects of the code that will make future improvements and maintenance easier. Some of the changes may have stronger impact in builds with lower optimization levels, such as is used by GROMACS developers or in its CI testing system. Together, the subsequent changes mitigated the performance loss from using std::optional, by lifting the logic that used it out of loops. It is worth noting that had these refactorings happened first, the impact on performance of introducing std::optional would likely have been negligible, which is a lesson for future changes.
Thanks also to the reviewers of these code changes, who all made useful contributions!
For all the past developments on GROMACS please follow this link https://enccs.se/tag/gromacs/.