The lead developer for the GROMACS Python API (gmxapi – see https://academic.oup.com/bioinformatics/article/34/22/3945/5038467), Dr Eric Irrgang of Prof. Peter Kasson’s lab (https://kassonlab.wordpress.com/), visited Stockholm recently, thanks to travel support from HPC-Europa3. Sadly the state of COVID-19 in the world meant that he was still distanced from many of the core GROMACS team in Stockholm. However Dr Mark Abraham of ENCSS was still able to use the time with Eric to focus on planning how to remove obstacles in the path of exascale-suitable API-driven molecular dynamics simulations.
The figure below depicts the kind of workflow we need to be able to generalize to run multiple ensemble members simultaneously.
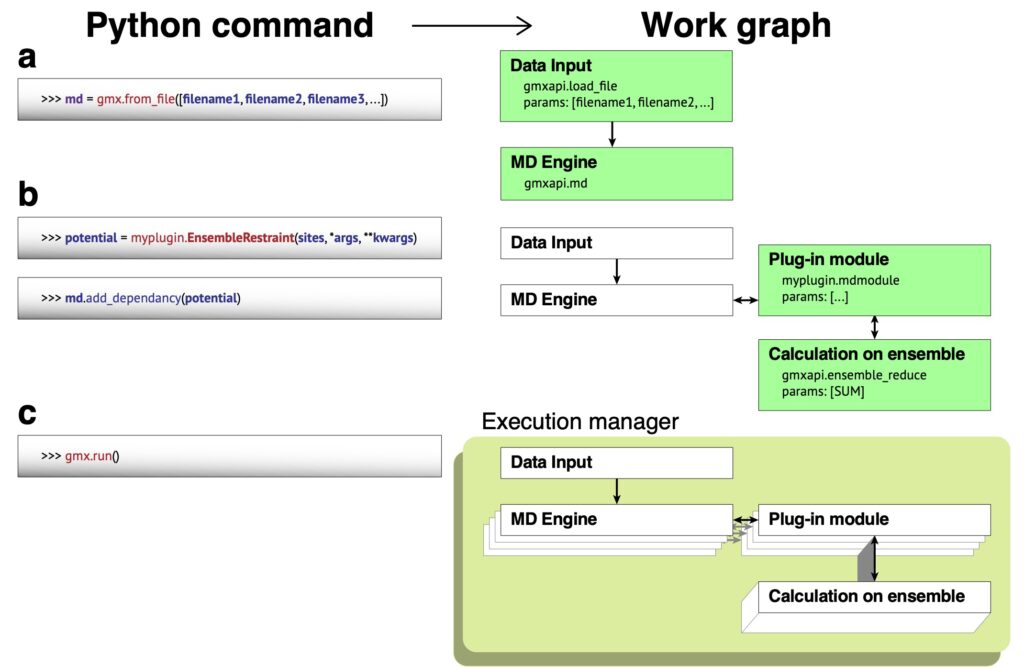
Like many HPC codes, GROMACS uses MPI to coordinate work across computer nodes within a simulation. However its history as a command-line program wasn’t the best preparation for being able to generalize the use of MPI across many simulations. Gmxapi could currently run multiple simulations, but there are severe restrictions on the amount of resources each simulation can address.
During the visit, Eric worked on infrastructure to allow the Python layer to manage an MPI context for running successive simulations, and Mark moved the GROMACS hardware detection to run earlier and inter-operate better with the external MPI context. Several awkward legacy code constructs were improved and the resulting code is much better suited for further work to support the generalization. Several related issues were observed during that work, and have been documented and planned for future resolution, e.g https://gitlab.com/gromacs/gromacs/-/issues/3774 and https://gitlab.com/gromacs/gromacs/-/issues/3688.
For more information on GROMACS visit: http://www.gromacs.org.
RECENT NEWS
[post_grid id=’651′]